有机基体/ 无机填料复合树脂是常用的牙科修复材料,但其主单体双酚A-双甲基丙烯酸缩水甘油酯(Bis-GMA)为双酚A 类单体,具有生殖毒性,长期接触会对人体造成不可逆伤害,并且易造成树脂聚合收缩率大的问题,易导致继发龋的产生。前期设计了一种新型桦木醇基树枝状单体(D1 )对Bis-GMA 进行取代,选用微米级钡玻璃粉作为无机填料,对钡玻璃粉进行硅烷化改性后按照质量分数24%、47%和71%与树脂基体共混制备复合树脂,通过旋转流变仪、红外光谱仪、万能试验机和密度天平等对复合树脂的性能进行探究。结果表明:相同填料量下,含有桦木醇基树枝状单体的复合树脂具有更优异的流变性,并且在5B5TM24 和D14B5TM24 的弯曲强度和弯曲模量分别为(84. 41±9. 08)MPa、(1. 86±0. 17)GPa 和(130. 50±9. 16)MPa、(2. 25±0. 23)GPa 的前提下,有效降低聚合收缩率,5B5TM71 为(4. 47 ± 0. 35)%,D14B5TM71 为(3. 52 ± 0. 23)%。因此,桦木醇基树枝状单体具有优异的性能,有潜力应用于临床使用的新型牙科修复材料。关键词: 桦木醇;复合树脂;树枝状单体;细胞相容性;聚合收缩率中图分类号: R783. 1 文献标识码: A 文章编号: 1006 334X(2025)02 0033 08文章来源:中国知网

龋病是一种最为普遍的慢性口腔细菌感染性疾病,不仅对牙齿的功能造成影响,还可能引发或加重多种疾病,例如关节炎、心内膜炎、慢性肾炎等[1] 。修复材料的选择直接影响龋齿的治疗效果。牙科修复材料的发展经历了从银汞合金向复合树脂的转变,目前商用的常用牙科修复材料中,双酚A 甘油二甲基丙烯酸酯(Bis-GMA)树脂体系使用最为广泛[2] 。但这类材料由于固化过程中发生聚合收缩现象,产生微渗漏,从而诱导继发龋,导致修复失败[3] 。此外,Bis-GMA 具有生殖毒性且为不可再生资源。为解决这一问题,国内外多个课题组已开展低收缩率单体的结构设计,例如开环聚合单体和超支化类的聚合物[4-5] 。这些结构的设计有效拓展了有机单体的种类,但相关合成步骤比较复杂[6-8] ,且固化时间较长,不利于实际应用。因此,设计开发一种新型桦木醇基树枝状单体,并将其应用于树脂基体中,以期降低聚合收缩率和细胞毒性。为进一步验证该类单体在复合树脂中的有效性,将表面硅烷化改性的钡玻璃粉作为无机填料与桦木醇基树枝状单体共混得到复合树脂,并对其流变性能、双键转化率、聚合收缩率、机械性能及生物相容性进行综合评估,为应用于临床牙科修复材料提供数据支持。1. 1 原料4-二甲氨基吡啶(DMAP),分析纯,上海麦克林生化科技有限公司; N, N - 二环己基碳二亚胺(DCC),分析纯,上海阿拉丁生化科技股份有限公司;2,2,5-三甲基-1,3-二恶烷-5-甲酸,分析纯,萨恩化学技术(上海) 有限公司;浓硫酸、樟脑醌(CQ)、4-二甲氨基苯甲酸乙酯 (4-EDMAB)、环己烷、正丙烷,分析纯,国药集团化学试剂有限公司;桦木醇(Betulin)、甲基丙烯酸酐、乙酸乙酯(EA)、二氯甲烷(DCM)、甲醇(MeOH),分析纯,阿达玛斯(上海)试剂有限公司;氢氧化钠、无水碳酸钠、氯化钠、无水硫酸镁,分析纯,上海泰坦科技股份有限公司;Bis-GMA、双甲基丙烯酸二缩三乙二醇酯(TEGDMA),γ-甲基丙烯酰氧基三甲氧基硅烷(γ-MPS),分析纯,西格玛奥德里奇(上海)贸易有限公司;钡玻璃粉,微米级(含硅量4. 6%),德国肖特公司。1. 2 仪器设备 双中心混合分散机,DAC 150. 1 FVZ-K 型,德国Hauschild 公司;三辊研磨机,80E型,德国EXAKT公司;红外光谱仪,Nicolet iS50 型,美国Nicolet公司;场发射扫描电子显微镜,SU 8010 型,日本日立公司;能谱仪,Quantax XFlash,德国Bruker 公司;安东帕高温旋转流变仪,Anton Paar MCR702 型,奥地利Anton Paar 公司; 电子万能材料试验机,NSTRON 33R 4201 型,美国Instron 公司;密度天平,AL/ AB-N 型,梅特勒-托利多仪器上海有限公司;热重分析仪,TG 209 F1 型,德国耐驰公司;酶标仪,Elx800 型, 美国Biotek 公司; 荧光显微镜, LeicaDMi8 型,德国徕卡公司。

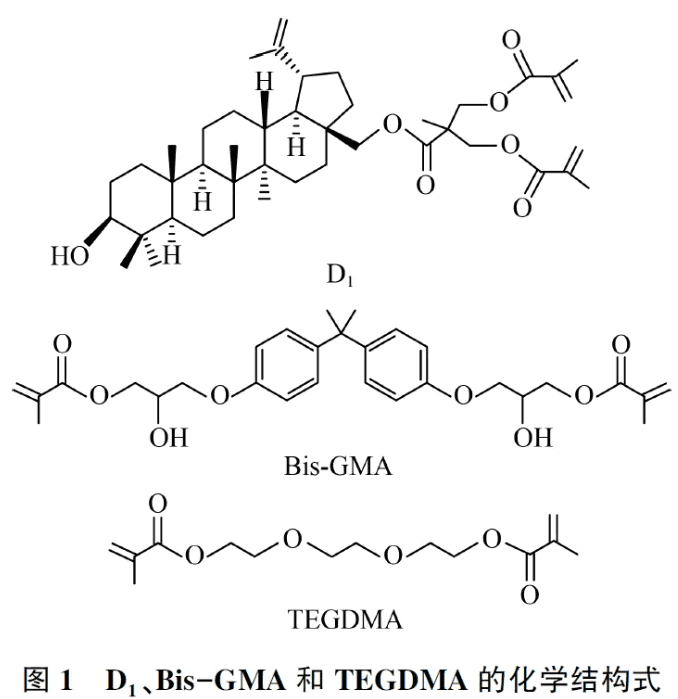
1. 3 实验过程 单体合成:在课题组前期工作基础上[9] ,以桦木醇为原料,通过对酯化反应的反应位点进行控制,制备了一系列末端含有反应性双键的树枝状单体,其中D1 单体与牙科修复树脂基体的相容性最佳,表现出优异的理化性能,优选D1 单体用于复合树脂的制备及相关测试,D1 及复合树脂中有机单体结构如图1 所示。
无机填料改性:称取5. 0 g 粒径为1 μm 的钡玻璃粉加入500 mL 圆底烧瓶中,加入100 mL 环己烷,搅拌均匀后分别加入0. 14 mL 丙胺和0. 53 mL γ-MPS,在60 ℃下回流反应1 h。待反应完成后,依次使用环己烷和乙醇洗涤3 次,将产物置于60 ℃真空烘箱中干燥24 h,得到白色粉末状产物,即表面硅烷化的钡玻璃粉(BGP-M)。复合树脂制备:称取5B5T 和D14B5T 作为树脂基体(5B5T:50% Bis-GMA 和50% TEGDMA 组成的树脂基体;D14B5T:10% D1、40% Bis -GMA 和50% TEGDMA 组成的树脂基体),分别选择质量分数为24%、47%和71%表面硅烷化改性后的钡玻璃粉作为无机填料,复合树脂的组成如表1 所示。在避光条件下采用双中心分散设备将无机填料与树脂基质进行预混合,随后将所得物置于三辊研磨机内混合均匀。 
1. 4 分析测试红外光谱测试:通过Nicolet 8700 型傅里叶红外光谱仪的衰减全反射附件ATR 测试钡玻璃粉改性前后的红外吸收光谱图,波数范围为650~4 000 cm-1,分辨率为4 cm-1,扫描次数为32 次。热失重测试:使用TG 209 F1 型热失重仪器检测钡玻璃粉的改性情况,在氮气吹扫气氛围下进行,升温速率为10 ℃ / min,温度区间为50~600 ℃。微观形貌测试:使用场发射扫描电子显微镜对改性前后钡玻璃粉的表面形貌进行分析。能谱测试:使用Quantax XFlash 型能谱仪在10~30 keV 范围内定量分析钡玻璃粉改性前后的元素组成与含量。剪切黏度测试:使用Anton Paar MCR702 型旋转流变仪在动态剪切模式下对复合树脂的流变性能进行测试,剪切范围为0. 1~100 s-1,温度为25 ℃。机械性能测试:使用INSTRON 33R 4201 型万能试验机参照ISO 4049—2019《牙科学 聚合物及充填、修复及粘接材料》测试,对复合树脂进行机械性能测试,通过万能试验机测量材料的弯曲强度和弯曲模量。细胞相容性测试:使用Elx800 型酶标仪和LeicaDMi8 型显微镜测量细胞相容性,根据标准ISO10993—5:2009《医疗器械的生物学评价第5 部分:细胞毒性试验体外方法》,将复合树脂样品制成直径为10 mm 的标准样品后,经紫外照射,乙醇洗涤杀菌处理后,制备25%、50%、75%和100%的浸提液,并与牙髓干细胞共培养24 h,通过CCK-8 和活/死细胞染色对细胞活性进行表征。1. 5 计算双键转化率:通过Nicolet 8700 型傅里叶红外光谱仪衰减全反射对复合树脂的双键转化率进行实时检测。脂肪族C =C 双键和C =O 在红外谱图中的吸收峰分别位于1 638 cm-1 和1 716 cm-1,根据公式(1)计算复合树脂的双键转化率。 式中h1 638 / h1 716 表示波数1 638 cm-1 处脂肪族C =C 双键和波数1 716 cm-1 处C =O 键吸收峰高度之比。
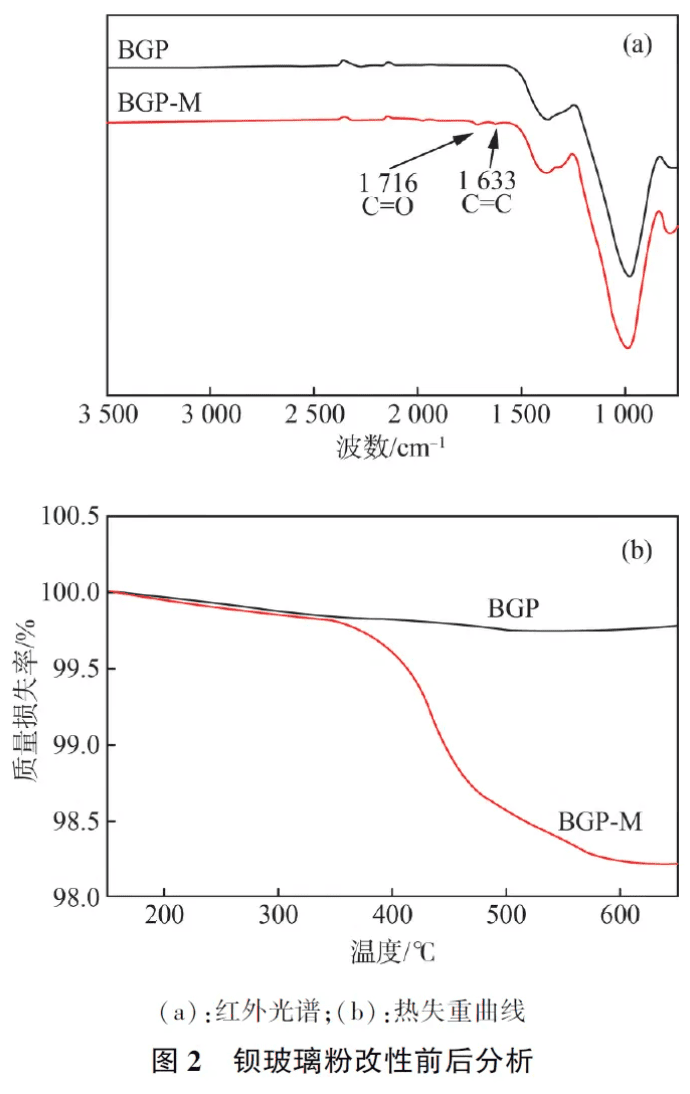
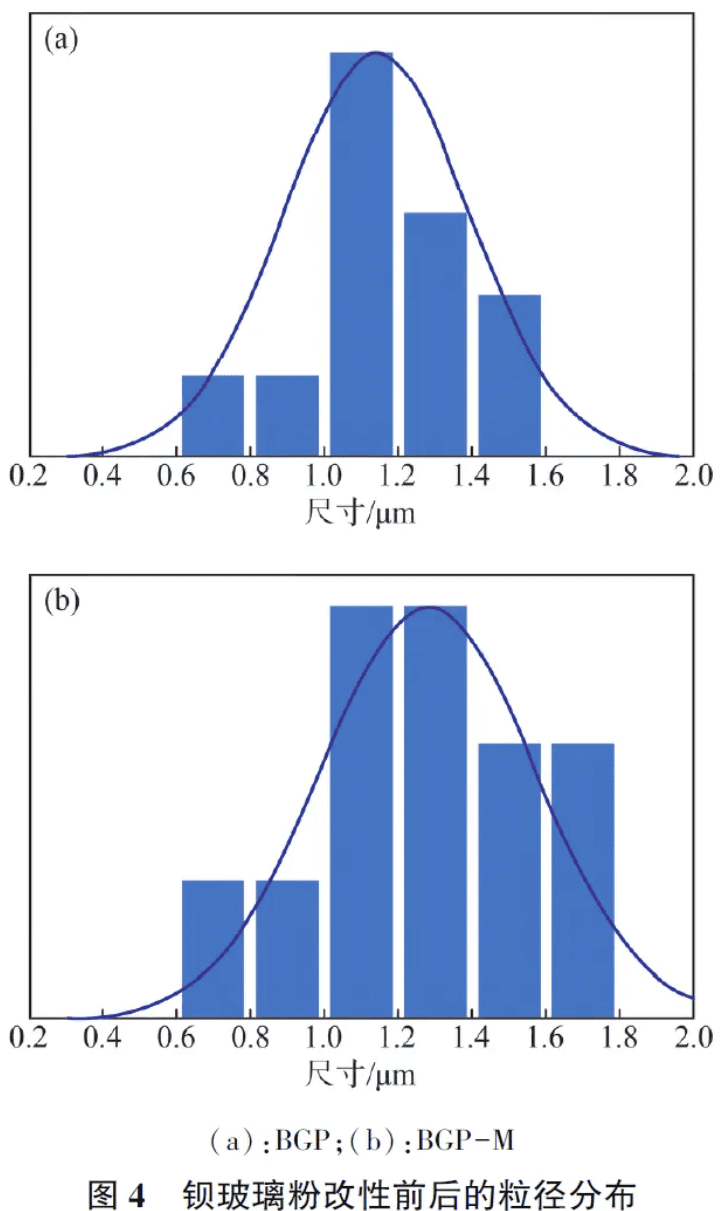
聚合收缩率:使用AL/ AB-N 型密度天平按照ISO 17304:2013《牙科学 聚合物基修复材料的聚合收缩的测定方法》对复合树脂聚合收缩率进行测试,通过阿基米德法测量聚合前后树脂的密度变化,根据公式(2)计算聚合收缩率。式中ρ固化和ρ未固化分别为固化前后复合树脂的密度。 2. 1 改性钡玻璃粉的结构表征在钡玻璃粉表面硅烷化改性的过程中,γ-MPS中甲氧基通过水解转化为Si—OH 键,并与填料表面的Si—OH 键发生缩合反应,生成Si—O—Si 的网状结构。这种结构覆盖于填料表面,并通过该反应将γ-MPS 的甲基丙烯酸酯官能团引入填料中,使得填料表面具有反应性C =C。首先,通过傅里叶红外光谱定性分析钡玻璃粉硅烷化前后的变化。如图2(a)所示,经表面改性后,在1 726 cm-1 和1 633 cm-1 处出现了γ-MPS 中的C =O 和C =C 吸收峰,证明钡玻璃粉表面硅烷化成功。为进一步定量分析钡玻璃粉硅烷化改性的程度,通过TGA 在50 ~ 600 ℃ 升温得到热失重曲线,如图2(b)所示。未改性钡玻璃粉在升温过程中无明显失重现象,最终质量损失率为0. 2%,这部分质量主要为钡玻璃粉表面的杂质和水分,而改性后的钡玻璃粉在350 ℃后出现明显失重,这是由于升温后表面硅烷偶联剂的分解,最终质量损失率为1. 8%,即为表面硅烷偶联剂的含量,证明已成功进行了硅烷化改性。此外,通过扫描电镜从微观上对钡玻璃粉的表面形貌进行分析,结果如图3 所示。通过FESEM图像可以看出,由于表面接枝的γ-MPS 分子量较小,改性前后钡玻璃粉的表面形貌无明显变化。利用Image J 对其粒径进行测量分析,如图4 所示, 结果表明BGP 的平均粒径为( 1. 14 ±0. 25)μm,改性后BGP -M 的粒径提升至(1. 28±0. 29)μm。随后对钡玻璃粉表面各元素进行分析,各元素含量如表2 所示,改性后钡玻璃粉表面C 元素含量由10. 27%提升至14. 64%,证明表面已引入γ-MPS 的碳链结构。
2. 1 改性钡玻璃粉的结构表征在钡玻璃粉表面硅烷化改性的过程中,γ-MPS中甲氧基通过水解转化为Si—OH 键,并与填料表面的Si—OH 键发生缩合反应,生成Si—O—Si 的网状结构。这种结构覆盖于填料表面,并通过该反应将γ-MPS 的甲基丙烯酸酯官能团引入填料中,使得填料表面具有反应性C =C。首先,通过傅里叶红外光谱定性分析钡玻璃粉硅烷化前后的变化。如图2(a)所示,经表面改性后,在1 726 cm-1 和1 633 cm-1 处出现了γ-MPS 中的C =O 和C =C 吸收峰,证明钡玻璃粉表面硅烷化成功。为进一步定量分析钡玻璃粉硅烷化改性的程度,通过TGA 在50 ~ 600 ℃ 升温得到热失重曲线,如图2(b)所示。未改性钡玻璃粉在升温过程中无明显失重现象,最终质量损失率为0. 2%,这部分质量主要为钡玻璃粉表面的杂质和水分,而改性后的钡玻璃粉在350 ℃后出现明显失重,这是由于升温后表面硅烷偶联剂的分解,最终质量损失率为1. 8%,即为表面硅烷偶联剂的含量,证明已成功进行了硅烷化改性。此外,通过扫描电镜从微观上对钡玻璃粉的表面形貌进行分析,结果如图3 所示。通过FESEM图像可以看出,由于表面接枝的γ-MPS 分子量较小,改性前后钡玻璃粉的表面形貌无明显变化。利用Image J 对其粒径进行测量分析,如图4 所示, 结果表明BGP 的平均粒径为( 1. 14 ±0. 25)μm,改性后BGP -M 的粒径提升至(1. 28±0. 29)μm。随后对钡玻璃粉表面各元素进行分析,各元素含量如表2 所示,改性后钡玻璃粉表面C 元素含量由10. 27%提升至14. 64%,证明表面已引入γ-MPS 的碳链结构。 
2. 2 复合树脂的流变性能剪切黏度是复合树脂的一项重要流变参数,与材料的双键转化率、吸水性和聚合收缩率等特性密切相关[10] ,因此,将钡玻璃粉与树脂基体复配得到复合树脂,首先探究其剪切黏度与剪切速率的关系。
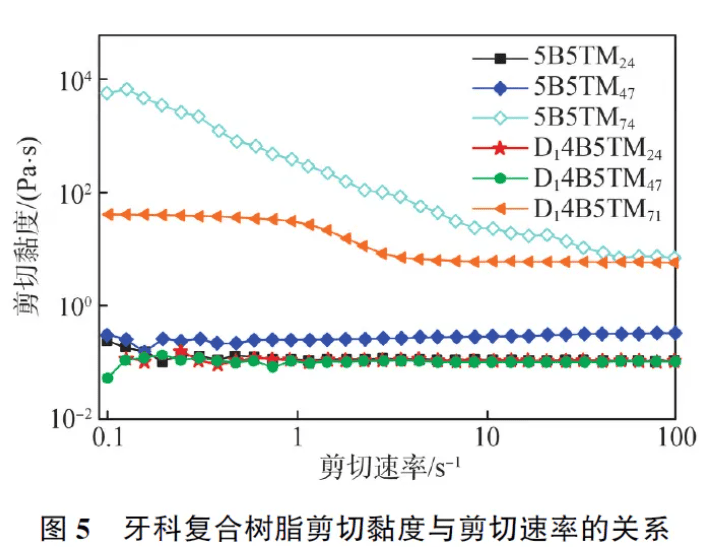
图5 为不同填料量的复合树脂剪切黏度随剪切速率的变化曲线,在相同填料量下,5B5T 树脂体系的剪切黏度高于D14B5T 体系,这是由于Bis-GMA 分子中的羟基含量高于D1,D1 的加入导致分子间作用力降低,复合树脂的剪切黏度降低[11] 。随着填料量的增加,各组均呈现剪切黏度快速上升的趋势,在最大填料量71%时,5B5TM71 和D14B5TM71 的剪切黏度分别为7. 31 Pa·s 和6. 15 Pa·s。这是由于无机填料呈固态,复合树脂的流动性主要由树脂基体提供,树脂基体中D1 单体的柔性链段有利于增强材料整体的流动性。
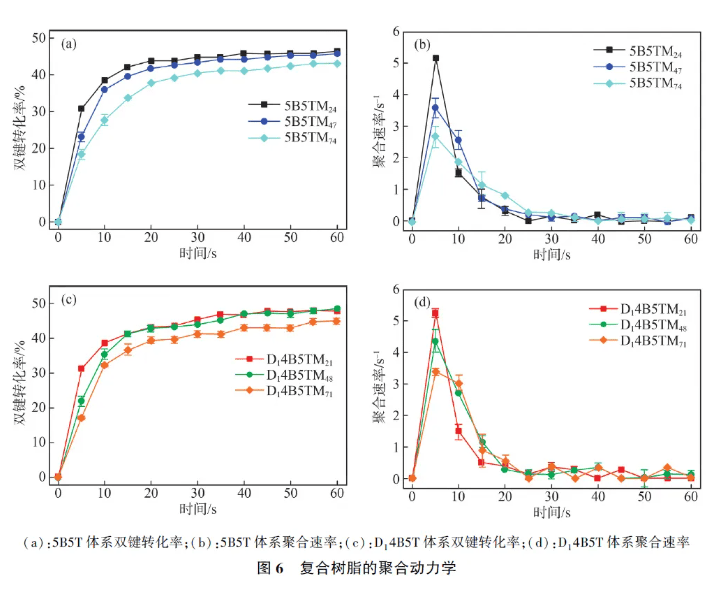
树脂基体较高的剪切黏度会导致无机填料的添加量降低,不利于加工和使用,并且由于高剪切黏度对无机相填料种类和添加量的限制[12] ,会降低材料的双键转化率、机械性能和使用寿命。虽然D1 单体为固体粉末,但是由于其较弱的分子间作用力和柔性链段,D14B5T 树脂基体在添加71%的钡玻璃粉后,剪切黏度仍能满足使用需求,其中剪切黏度最佳的实验组为D14B5TM24。2. 3 复合树脂的双键转化率牙科复合材料的固化机制涉及树脂基体与无机填充材料中的双键相互作用,通过聚合反应形成交联网络结构。在这一过程中,双键转化率被视为评估光固化反应进展的关键参数。图6(a)和图6(c)分别为5B5T 和D14B5T 树脂基体中添加24%、47%和71%的BGP-M 时,通过实时红外光谱计算出的60 s内实时双键转化率曲线。通过观察数据可知,光照开始后的最初20 s 内,不同实验组的双键转化率均呈现快速增长趋势,此时为聚合初期的自动加速现象[13] 。当照射时间超过20 s 后,各组的双键转化率基本趋于稳定,继续延长光照时间仅能引起极微小的变化,最终双键转化率均维持在43%至49%之间。由于双键转化率的提高,复合树脂的剪切黏度随之增加,这在一定程度上阻碍了未聚合单体的移动和链增长过程。随着剪切黏度的进一步增加,反应也随之停止,残余的双键也无法继续固化。5B5T树脂体系随着填料量的增加,复合树脂的双键转化率由(45. 24±0. 35)%降低至(43. 01±0. 28)%。同样的,D14B5T 的双键转化率也随着填料量的增加而降低,这归因于钡玻璃粉的主要成分为无机物,仅有表面含有接枝的双键可以参与聚合,导致复合树脂的整体双键浓度降低。另一方面,无机填料的加入降低了复合树脂的流动性,不利于链段的移动。D14B5T 在填料量为24%和47%时,其双键转化率分别为(45. 68±0. 50)%和(44. 22±0. 52)%,并未出现明显下降,这得益于复合树脂较好的流动性及树脂基体中更高的双键浓度。 进一步计算双键转化率图像的斜率,即可得到复合树脂的聚合速率图,如图6(b)和6(d)所示,由图可知,所有复合树脂在5 s 左右均达到最大聚合速率,5B5TM24 为(5. 16 ± 0. 05) s-1,D14B5TM24 为(5. 24±0. 12)s-1。随着填料量的增加,聚合速率也随之降低。树脂基体中加入填料后的透光率降低, 所以整体的聚合速率也降低,但在D14B5T 基体中,随着填料的增加,复合树脂的双键转化率呈现先上升再下降的趋势,这可能是由于D14B5T 基体的剪切黏度较低,与钡玻璃粉之间的相容性更好,填料量不高时两相均匀混合有利于树脂基体在无机填料表面进行快速聚合。在各实验组中, 5B5TM24 和D14B5TM24 具有最佳的转化率和聚合速率。
2. 4 复合树脂的机械性能图7 和图8 分别为不同填料量的复合树脂的弯曲强度和弯曲模量。填料的加入显著增强了树脂基体的力学性能,γ-MPS 与树脂基体共聚,使得有机相和无机相结合,增强了两相间作用力[14] 。
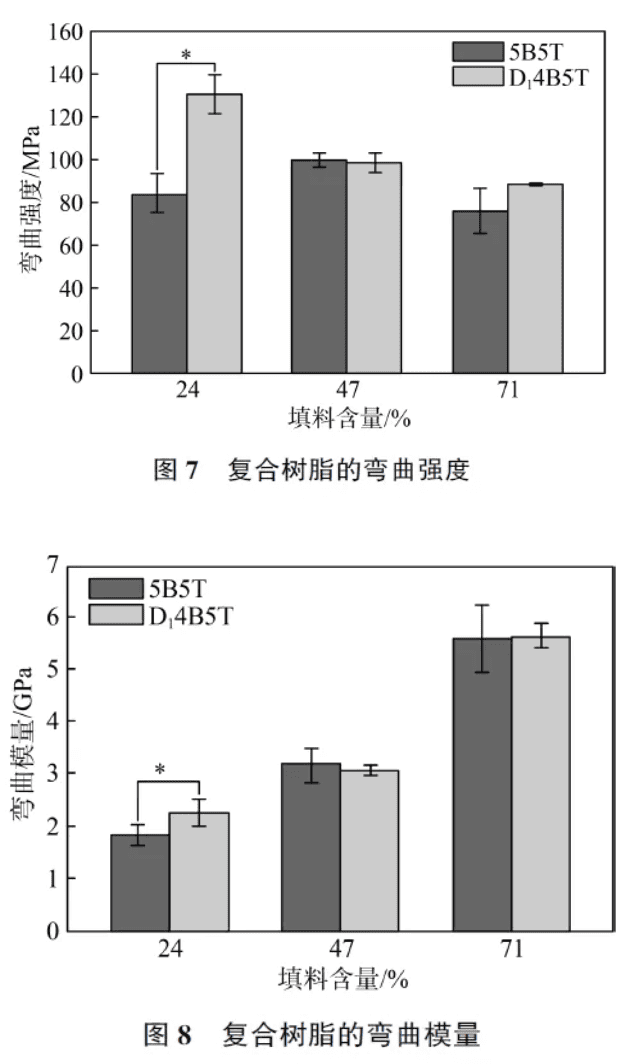
其中,D14B5TM24 的弯曲强度和弯曲模量分别为( 130. 50 ± 9. 16) MPa 和( 2. 25 ± 0. 23) GPa,5B5TM24 的弯曲强度和弯曲模量分别为(84. 41±9. 08)MPa 和(1. 86±0. 17)GPa,两者间存在显著性差异,随着填料量进一步增加,两者间的差异逐渐缩小。当填料量较低时,复合树脂主要体现树脂基体的性能, 此时D14B5TM24 的双键转化率高于5B5TM24,有利于增强复合树脂的弯曲强度和弯曲模量,填料量较高时则会削弱两者间的差异。
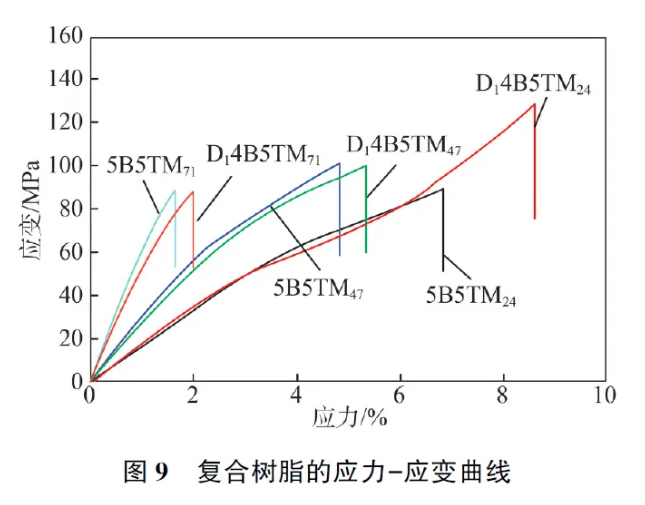
图9 为树脂基体在拉伸过程中的应力-应变曲线,填料量高于47%时,5B5T 与D14B5T 的力学行为较为接近,此结果与材料的弯曲性能保持一致。此外,对应力-应变曲线所包围的面积进行积分,可计算出各树脂基体的断裂能[15] ,如图10 所示。
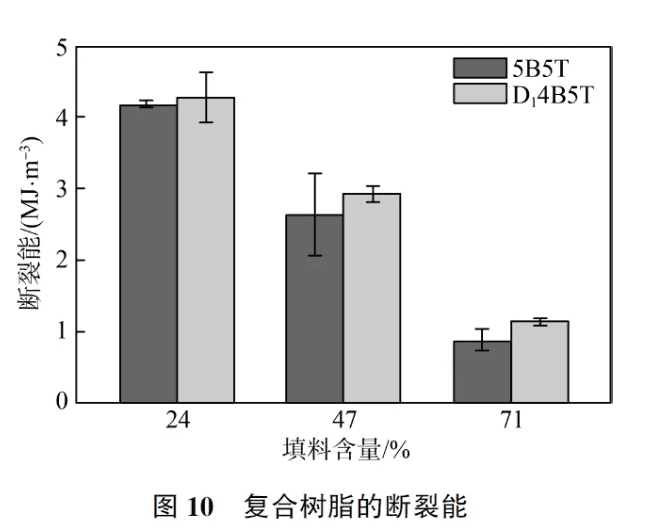
由图10 可知,填料的加入虽然提高了材料的力学强度,同时也降低了材料的韧性,含有柔性链D1 的实验组韧性会改善这一现象。以上结果证明D1 单体的适量加入不会削弱复合树脂的机械性能,其中D14B5TM24 的机械性能最佳。
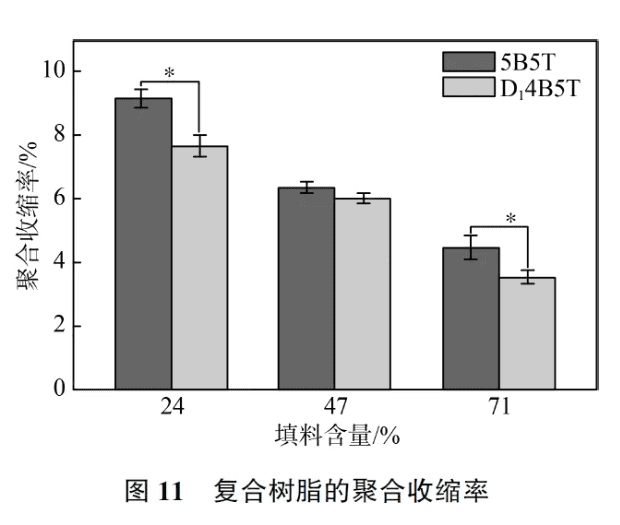
2. 5 复合树脂的聚合收缩率光固化复合树脂在使用过程中通常存在体积收缩的问题, 从而导致边缘渗漏和界面间隙等问题[16] 。图11 为不同填料量下复合树脂的聚合收缩率,随着填料量的增加,复合树脂的聚合收缩率显著降低[17-18] , 5B5TM24 从( 9. 12 ± 0. 28)% 降低至(4. 47±0. 35)% ,D14B5TM24 从(7. 64±0. 31)%降低至(3. 52±0. 23)%。填料的增加会大幅降低复合树脂中的双键浓度,从而通过减少聚合以缩小体积收缩率[19] 。同时,在加入填料后,含树枝状单体的实验组仍保持着更低的聚合收缩率。这是由于树枝状单体的分子量和分子体积高于Bis - GMA 和TEGDMA,导致单位体积树脂基体的C =C 双键数量较少[20] 。此外,弹性形变会伴随着较高的聚合收缩率与应力释放,D14B5T 体系的剪切黏度低于5B5T,有利于减少聚合过程中的弹性形变和聚合收缩率,D14B5TM24 与相同填料量下的对照组相比有显著的降低聚合收缩率的效果。
2. 6 复合树脂的细胞毒性选取综合性能较优的D14B5TM24 进行细胞毒性试验,5B5TM24 为对照组,图12 为5B5TM24 和D14B5TM24 在不同浓度浸提液对牙髓干细胞培养24 h 后,测得的牙髓细胞在450 nm 波长下的吸收值。
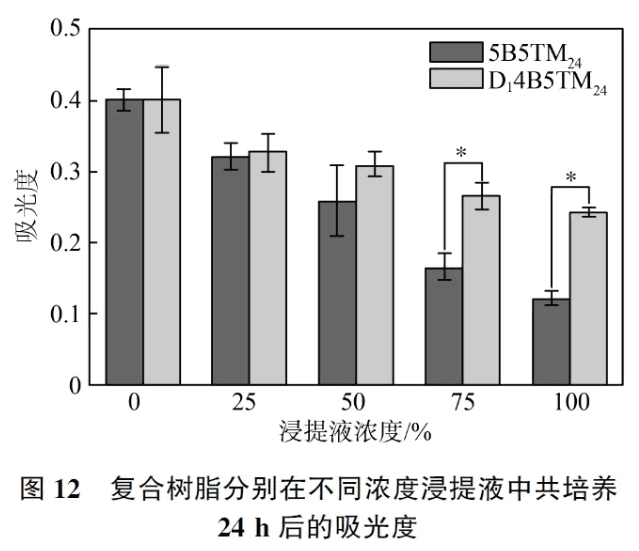
由图12 可知,牙髓干细胞在无浸提液的培养基中培养24 h 后的吸光度为0. 4,与对照组相比,将浸提液的浓度稀释至50%及以下时,未呈现明显的细胞毒性, 而在75% 和100% 的浓度下,D14B5TM24 的细胞毒性显著低于5B5TM24。由于单体难以完全聚合,复合树脂中会有残留的有毒单体,浸泡后在溶液中对细胞造成损伤,浸提液浓度越高这一现象越明显,但D14B5TM24 的细胞毒性随着浓度升高增强较慢,主要原因是:桦木醇作为天然生物基分子[21] ,细胞毒性低于Bis-GMA,D14B5TM24 的交联密度和双键转化率均高于5B5TM24,使得复合树脂中具有更少的未聚合单体。这表明桦木醇基树枝状单体在牙科树脂领域有潜在的应用前景。 将桦木醇基树枝状单体D1 对Bis-GMA 进行部分取代后,与改性后的微米级钡玻璃粉进行共混制得复合树脂。a) 与传统复合树脂5B5T相比,D14B5TM71聚合收缩率显著降低,为(3. 52±0. 23)%。此外,含桦木醇的树脂体系细胞相容性更为优异,在临床应用中具有优势。b) D1有潜力取代Bis-GMA 应用于牙科修复树脂。并且本研究也为高强度、低收缩以及低细胞毒性的牙科树脂单体结构设计提供了思路。考虑到口腔环境的复杂性,此类新型牙科修复树脂仍需进一步研究验证其实用性。
将桦木醇基树枝状单体D1 对Bis-GMA 进行部分取代后,与改性后的微米级钡玻璃粉进行共混制得复合树脂。a) 与传统复合树脂5B5T相比,D14B5TM71聚合收缩率显著降低,为(3. 52±0. 23)%。此外,含桦木醇的树脂体系细胞相容性更为优异,在临床应用中具有优势。b) D1有潜力取代Bis-GMA 应用于牙科修复树脂。并且本研究也为高强度、低收缩以及低细胞毒性的牙科树脂单体结构设计提供了思路。考虑到口腔环境的复杂性,此类新型牙科修复树脂仍需进一步研究验证其实用性。[1] 杨俊杰, 王亚子, 王瑞莉, 等. 有机-无机杂化齿科修复树脂材料的研究与发展前景[J]. 中国材料进展, 2015, 34(6):444-452.
[2] ALTIN A, AKGUN B, BUYUKGUMUS O, et al. Synthesis and photopolymerization of novel, highly reactive phosphonated-urea- methacrylates for dental materials [J]. Reactive and Functional Polymers, 2013, 73(9): 1319-1326.
[3] YU O Y, GE K X, CHU C H. The preventive effect of glass ionomer cement restorations on secondary caries formation: A systematic review and meta analysis [ J]. Dental Materials. 2023, 39 (12):e1-e17.
[4] ZHOU Z, LI A, SUN K, et al. Synthesis of a novel monomer“DDTU-IDI” for the development of low shrinkage dental resin composites[J]. Dental Materials. 2024, 40: 608-618.
[5] MATURI M, SPANU E, MACCAFERRI E, et al. (Meth)acrylate- free three-dimensional printing of bio-derived photocurable
resins with terpene-and itaconic acid-derived poly ( ester thioether) s [ J]. ACS Sustainable Chemistry & Engineering. 2023, 11(49): 17285-17298.
[6] CATEL Y, ANGERMANN J, FASSLER P, et al. High refractive indexmonofunctional monomers as promising diluents for dental composites [J]. Dental Materials, 2021, 37(2): 351-358.
[7] PéREZ-MONDRAGóN A A, CUEVAS-SUáREZ C E, CASTILLO O R S, et al. Evaluation of biocompatible monomers as substitutes for tegdma in resin-based dental composites[ J]. Materials Science and Engineering: C, 2018, 93(7): 80-87.
[8] CUEVAS-SUAREZ C E, GONZALEZ-LOPEZ J A, DA SILVA A F, et al. Synthesis of an allyl carbonate monomer as alternative to TEGDMA in the formulation of dental composite resins [J]. Journalof the Mechanical Behavior of Biomedical Materials, 2018, 87(7): 148-154.
[9] QIANG H J, WANG J J, WANG R L, et al. Plant-derived multimethacrylate dendritic monomers: Synthesis and applications in dental restorative resins[J]. Bioorganic Chemistry, 2025, 161 (7): 108552.
[10] DAVY K W, KALACHANDRA S, PANDAIN M S, et al. Relationship between composite matrix molecular structure and properties [J]. Biomaterials, 1998, 19(22): 2007-2014.
[11] SRIVASTAVA R, WOLSKA J, WALKOWIAK-KULIKOWSKA J, et al. Fluorinated bis-GMA as potential monomers for dental restorative composite materials [J]. European Polymer Journal, 2017,90(3): 334-343.
[12] HABIB E, WANG R, ZHU X X. Monodisperse silica-filled composite restoratives mechanical and light transmission properties [J]. Dental Materials, 2017, 33(3): 280-287.
[13] DAVIDENKO N, GARCIA O, SASTRE R. Photopolymerization kinetics of dimethacrylate-based light-cured dental resins [J]. Journal of Applied Polymer Science, 2005, 97(3): 1016-1023.
[14] DU M, ZHENG Y. Modification of silica nanoparticles and their application in UDMA dental polymeric composites [J]. Polymer Composites, 2007, 28(2): 198-207.
[15] WU X R, SUN Y, XIE W L, et al. Development of novel dental nanocomposites reinforced with polyhedral oligomeric silsesquioxane (POSS) [J]. Dental Materials, 2010, 26(5): 456-462.
[16] WATTS D C, KISUMBI B K, TOWORFE G K. Dimensional changes of resin/ ionomer restoratives in aqueous and neutral media [J]. Dental Materials, 2000, 16(2): 89-96.
[17] WANG Z Z, MARTIN Y M. System compliance dictates the effect of composite filler content on polymerization shrinkage stress [J]. Dental Materials, 2016, 32(4) : 551-560.
[18] BARNES D M, BLANK L W, GINGELL J C, et al. A clinical evaluation of a resin modified. glass ionomer restorative material
[J]. Journal of the American Dental Association, 1995, 126(9): 1245-1253.
[19] LU H, LEE Y K, OGURI M, et al. Properties of a dental resin composite with a spherical inorganic filler [J]. Operative Dentistry, 2006, 31(6): 734-740.
[20] DU M H, ZHENG Y. Modification of silica nanoparticles and their application in UDMA dental polymeric composites[J]. Polymer Composites, 2007, 28(2):198-207.
[21] ZHANG L S, MA Z Y, WANG R L, et al. Synthesis and characterization of methacrylate-functionalized betulin derivatives as antibacterial comonomer for dental restorative resins[J]. ACS Biomaterials Science & Engineering, 2017, 7(7): 3132-3140.



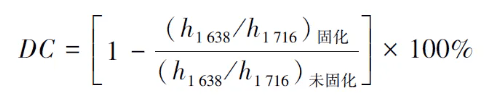
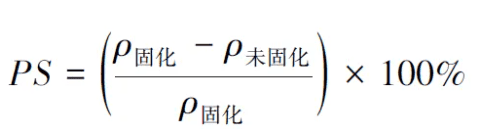


 扫一扫 关注我们
扫一扫 关注我们